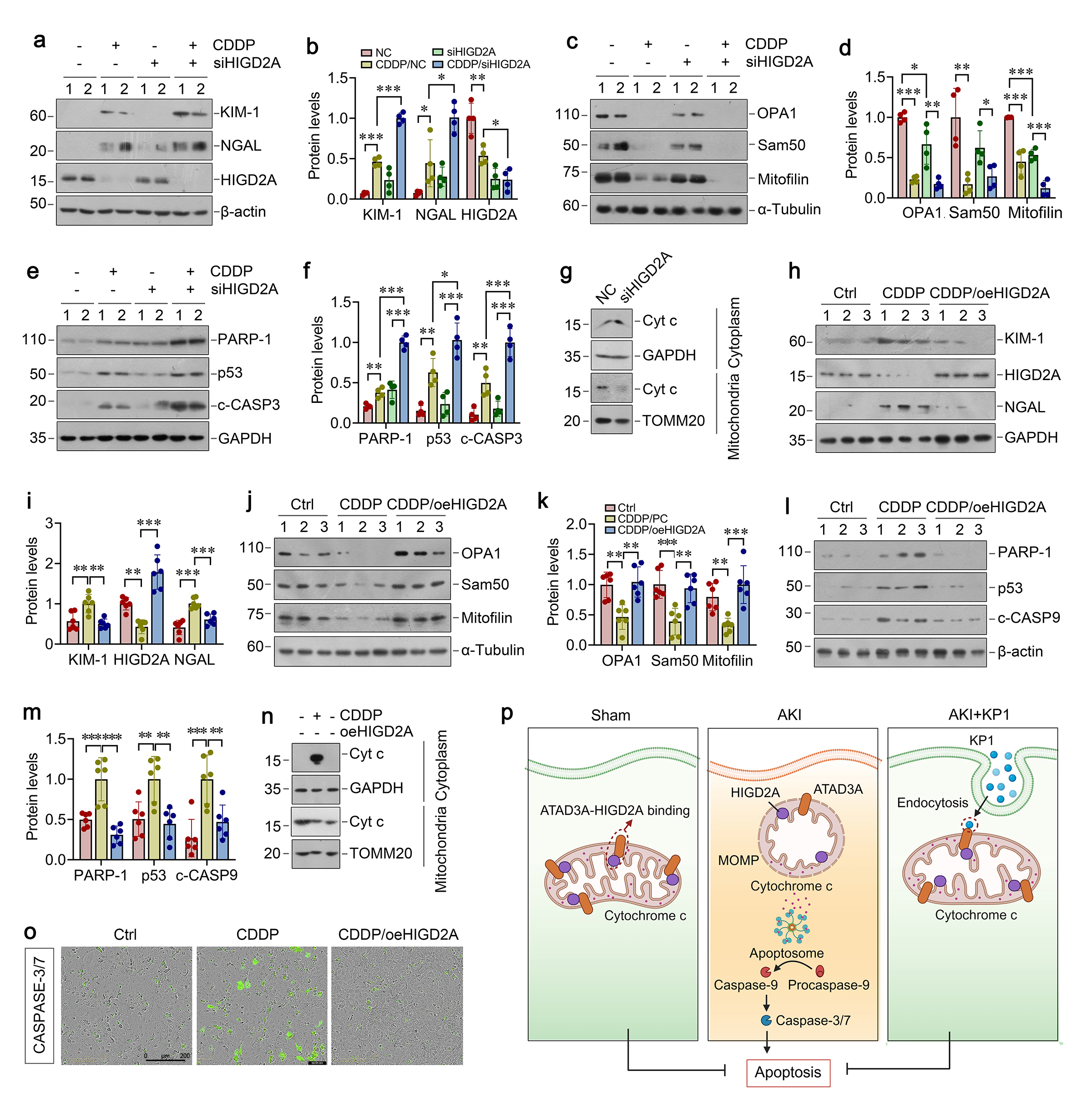
KP1 protects against both nephrotoxic and ischemic AKI in mice
We first examined the potential role of KP1 in AKI induced by cisplatin (CDDP), a widely used nephrotoxic drug. As shown in Figure 1a, Scr and BUN assays revealed that KP1 ameliorated CDDP-induced kidney dysfunction. KP1 attenuated CDDP-induced upregulation of renal injury markers kidney injury molecule-1 (Kim-1) and neutrophil gelatinase-associated lipocalin (NGAL) (Figure 1b and c), apoptosis markers poly(ADP-ribose) polymerase 1 (PARP-1), p53, cleaved caspase-3 (Figure 1d and e). Periodic acid-Schiff (PAS) staining revealed that KP1 prevented CDDP-induced pathohistological alterations, including tubular epithelial flattening, cell death, basement membrane rupture, tubular dilation, and prominent cast formation (Figure 1f). TUNEL staining also demonstrated that KP1 largely abolished tubular cell apoptosis induced by CDDP in the kidneys (Figure 1f). We found that CDDP-induced AKI was associated with depletion of mitochondrial proteins OPA1, Sam50, mitofilin, which were almost completely restored by KP1 (Figure 1g and h), suggesting a special role of KP1 in protecting against mitochondrial dysfunction in AKI.
To generalize these findings, we employed ischemic AKI induced by bilateral ischemia/reperfusion injury (IRI). As shown in Figure 1i, KP1 also reduced IRI-induced Scr and BUN levels. KP1 abolished IRI-upregulated Kim-1, NGAL, PARP-1, p53, and cleaved caspase-3 (Figure 1j-m). Histologically, KP1 prevented IRI-induced pathological alterations and reduced apoptosis in kidney tubular epithelium (Figure 1n). Notably, KP1 conferred mitochondrial protection by restoring IRI-depleted mitochondrial proteins OPA1, Sam50, and mitofilin (Fig. 1o and p). Electron microscopy also confirmed that KP1 preserved the ultrastructural integrity of mitochondria in tubular cells. KP1 reestablished healthy mitochondria with ordered cristae and elongated morphology by preventing IRI-induced fragmented and spherical transformation, membrane blurring, and cristae disorganization (Figure 1n).
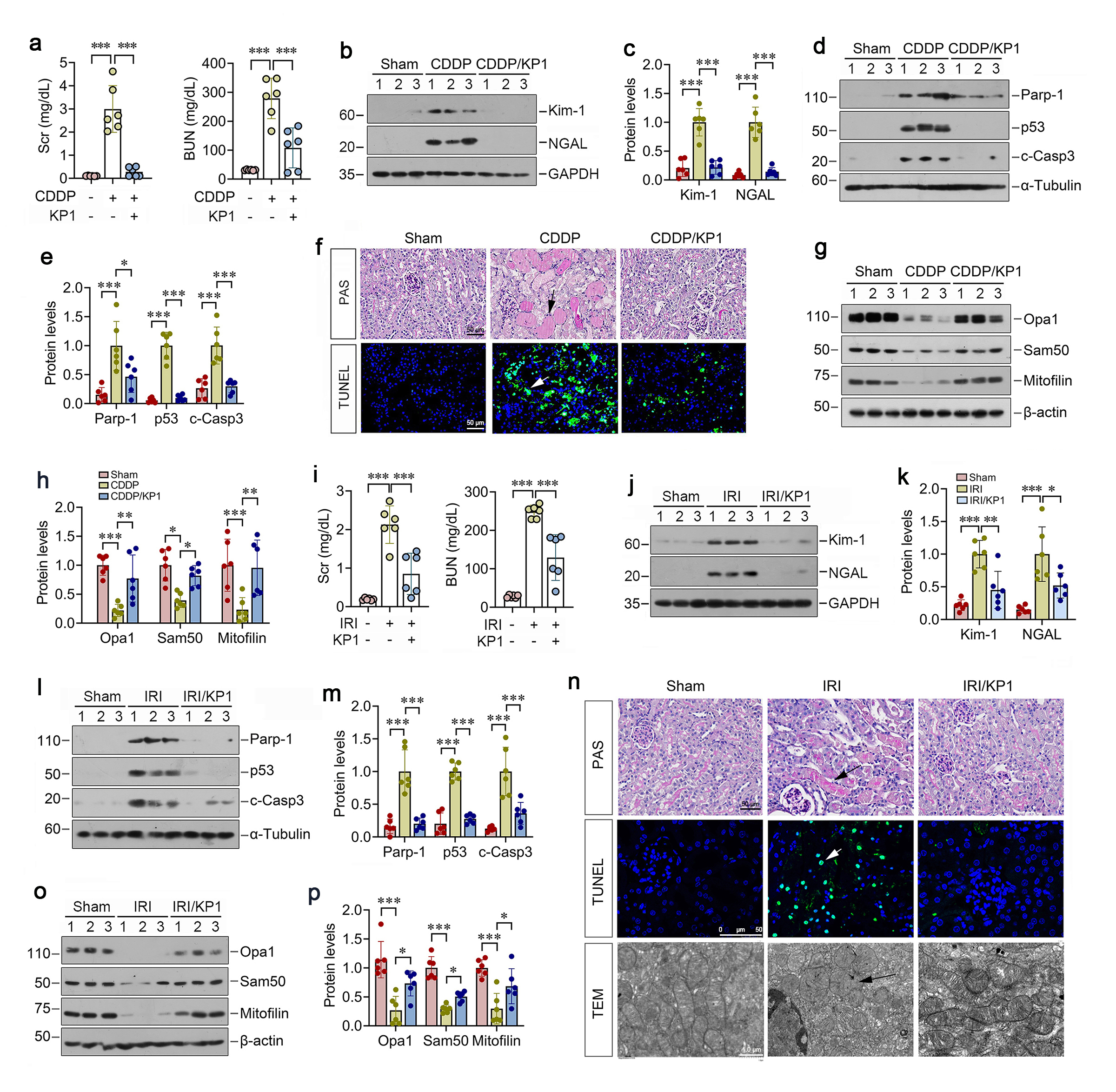
KP1 prevents renal tubular injury and mitochondrial dysfunction in vitro
We next investigated the role of KP1 in protecting tubular cells against nephrotoxic and ischemic injury in vitro. As shown in Figure 2a-d, CDDP induced the upregulation of tubular injury markers Kim-1 and NGAL, apoptosis mediators PARP-1, p53, and cleaved caspase-3 in HKC-8 cells, which were abolished by KP1. Longitudinal high-throughput live cell imaging also revealed that KP1 time-dependently suppressed CDDP-triggered caspase-3/7 cleavage and activation after 24 h of incubation in HKC-8 cells (Figure 2e and f). Furthermore, KP1 restored mitochondrial proteins OPA1, Sam50 and mitofilin in CDDP-treated HKC-8 cells (Figure 2g and h). Stimulated emission depletion (STED) microscopy revealed an imaging of mitochondria with nanoscale resolution and showed elongated mitochondria with fine cristae in live tubular epithelial cells. However, CDDP induced dramatic transformation of mitochondrial structure, with smaller round shape due to increased fission and disorganized cristae (Figure 2i). Quantitative determination showed that CDDP treatment reduced the size of mitochondria and the ratio of its length to its width (Figure 2j), while KP1 could restore all of these (Figure 2i and j). Label free live cell microscopy imaging showed that induced by CDDP mitochondria became small and short after 18 h with or without KP1treatment (Figure 2k). Mitochondria became much smaller and shorter without KP1 after 36 h, while recovered elongated with KP1 treatment (Figure 2k).
We also assessed the role of KP1 in protecting tubular cells against hypoxic injury by using a hypoxia/reoxygenation (H/R), an in vitro model imitating IRI in vivo. KP1 also reversed the ultrastructural changes of mitochondria triggered by H/R, as it alleviated mitochondria spherical transformation and cristae disorganization (Figure 2l). As shown in Figure 2n-q, KP1 inhibited H/R-induced upregulation of Kim-1, NGAL, PARP-1, p53, and cleaved caspase-3 in proximal tubular HKC-8 cells. Concomitantly, KP1 preserved mitochondrial integrity by restoring H/R-depleted OPA1, Sam50, mitofilin (Figure 2r and s).
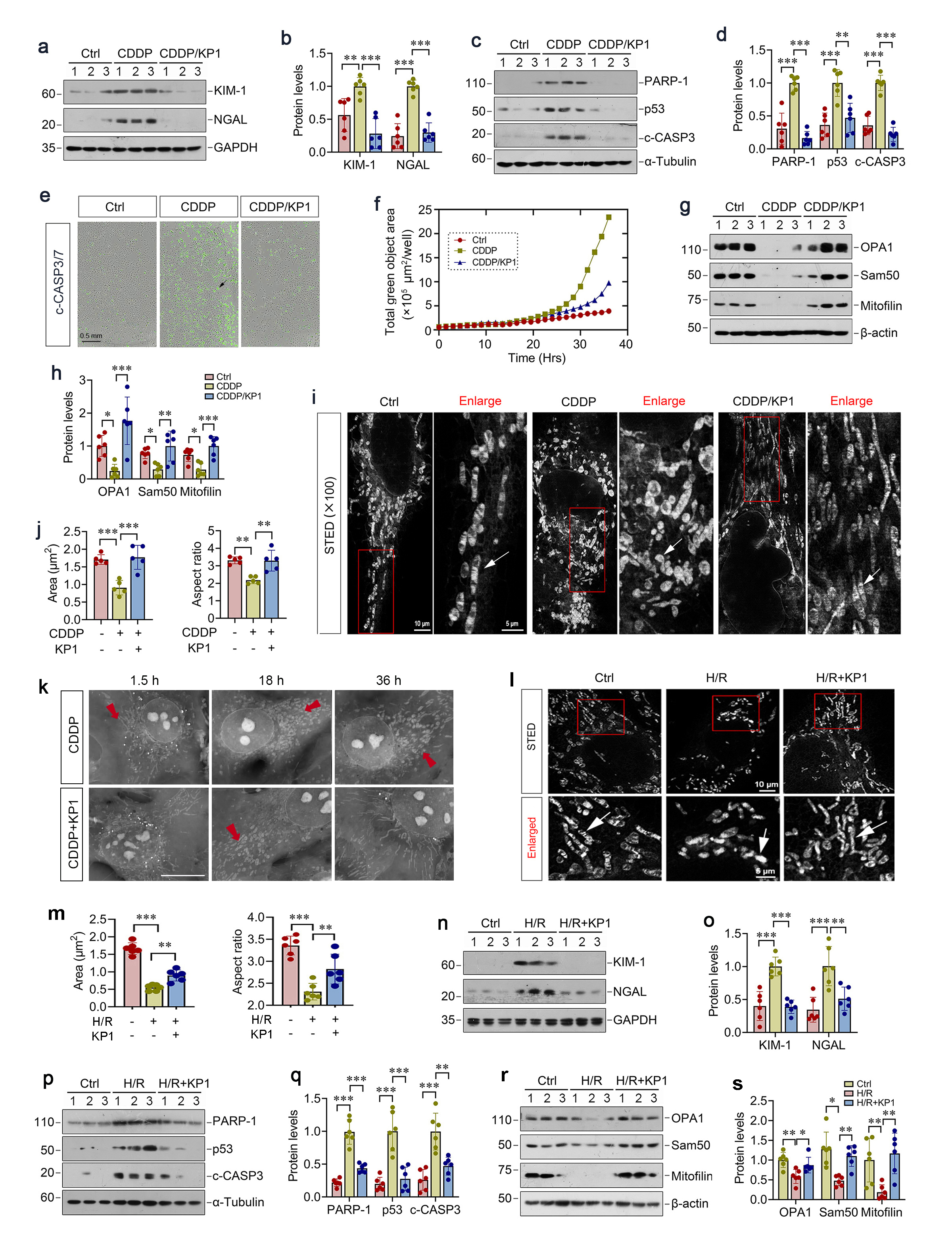
KP1 elicits its protection by entering into tubular cells via endocytosis
KP1 has been reported to interact with TGF-β receptor II (TβR2) and inhibit its downstream signaling in CKD. However, we found that CDDP and IRI did not significantly alter p-Smad3/Smad3 or TβR2 levels, which were also unaffected by KP1. Therefore, these results suggest that KP1-mediated renoprotection following AKI is independent of regulating TGF-β/Smad3 signaling.
To elucidate the mechanisms by which KP1 protects against AKI, we first sought to investigate how KP1 elicits its activity by crossing plasma membrane. To this end, we incubated HKC-8 cells with FITC-KP1 to trace its localization, and observed a time-dependent cytoplasmic accumulation of FITC-KP1 (Figure 3a). Using longitudinal live-cell imaging and pHrodo-Red Dextran, we confirmed FITC-KP1 co-localization with endocytic vesicles at 3 h, whereas co-staining unconjugated FITC and pHrodo-Red was negligible (Figure 3b), indicating endocytosis-dependent uptake of KP1 by HKC-8 cells. Furthermore, incubation with endocytosis inhibitor prozaleazine (PCZ) clearly blocked FITC-KP1 entry to HKC-8 cells, as assessed by flow cytometry (Figure 3c).
We found that blockade of KP1 uptake by PCZ abolished the cytoprotective activity of KP1 following CDDP treatment. In the presence of PCZ, KP1 was unable to inhibit Kim-1, NGAL, PARP-1, p53, cleaved caspase-3 induction by CDDP (Figure 3d-g). Similarly, blockade of KP1 uptake by PCZ also abolished the ability of KP1 to restore mitochondrial proteins OPA1, Sam50, and mitofilin in HKC-8 cells (Figure 3h and i). These results indicate that the reno-protective role of KP1 is dependent on its entry into tubular epithelial cells.
To closely imitate the in vivo situation, we utilized primary mouse proximal tubular epithelial cells, characterized by E-cadherin⁺/vimentin⁻ immunostaining (Figure 3j), to assess the ability of KP1 to protect tubular cells against nephrotoxic or hypoxic injury. As shown in Figure 3k and l, KP1 suppressed CDDP-induced apoptosis in primary tubular cells, as assessed by flow cytometry. KP1 also repressed the expression of PARP-1, p53, and cleaved caspase-3 (Figure 3m and n). KP1 concurrently reduced injury marker NGAL and restored mitochondrial proteins OPA1 and Sam50 in primary tubular cells (Figure 3o-p). Similarly, KP1 was able to reduce cell apoptosis triggered by H/R (Figure 3q), inhibited H/R-induced PARP-1, p53, cleaved caspase-3 and NGAL expression, and restored mitochondrial OPA1 and mitofilin proteins after H/R treatment (Figure 3r-u).
KP1 directly binds mitochondrial protein ATAD3A
To identify intracellular targets of KP1, we employed co-immunoprecipitation coupled with mass spectrometry (Co-IP/MS) approach. HKC-8 cells were incubated with FITC or FITC-KP1, and underwent immunoprecipitation using anti-FITC-conjugated agarose beads, followed by analyzing with mass spectrometry (Figure 4a). Co-IP/MS revealed that KP1 interacted with multiple mitochondrial proteins. Among them, the most prominent one was ATAD3A (Figure 4b). Molecular docking simulation revealed that KP1 exhibited high affinity to ATAD3A, with G14, Y18 and Q19 as contact sites, which corresponded to Gly70, Tyr74 and Gln75 in human αKlotho protein, respectively (Figure 4c and d). Furthermore, Co-IP analysis demonstrated that FITC-KP1 physically interacted with ATAD3A in HKC-8 cells (Figure 2C and D), validating the binding of KP1 to ATAD3A. Point mutations of KP1 by substituting with alanine at the predicted binding residues G14, Y18, and Q19 abolished KP1/ATAD3A interaction in Co-IP assays (Figure 4e). Confocal microscopic imaging confirmed the intracellular co-localization of KP1 and ATAD3A (Figure 4f). To further ascertain mitochondrial localization of KP1, we employed immunogold electron microscopy localize FITC-KP1 and visualized its presence to mitochondrial membranes in HKC-8 cells (Figure 4g), Microscale thermophoresis (MST), an immobilization-free and fluorescence-based biophysical analysis of interactions between biomolecules, confirmed high-affinity binding between KP1 and ATAD3A, with Kd = 3.19×10-7 M (Figure 4h and i).
To investigate the sequel of KP1 binding to ATAD3A, we utilized the drug affinity responsive target stability (DARTS) assays (Figure 4j). As shown in Figure 4k-l, KP1 dose-dependently protected ATAD3A from pronase-mediated degradation, indicating that KP1 stabilizes its target protein ATAD3A upon binding. We found that in CDDP or IRI models, KP1 treatment completely prevented injury-induced ATAD3A depletion (Figure 4m-q). Furthermore, tubular cell injury induced by CDDP or H/R caused depletion of ATAD3A protein in cultured HKC-8 cells, which was restored by KP1 incubation (Fig. 4r and s). Collectively, these results indicate that KP1 can directly binds to mitochondrial protein ATAD3A and stabilizes it by preventing injury-induced degradation.
ATAD3A preserves mitochondrial functions and protects against AKI in vivo
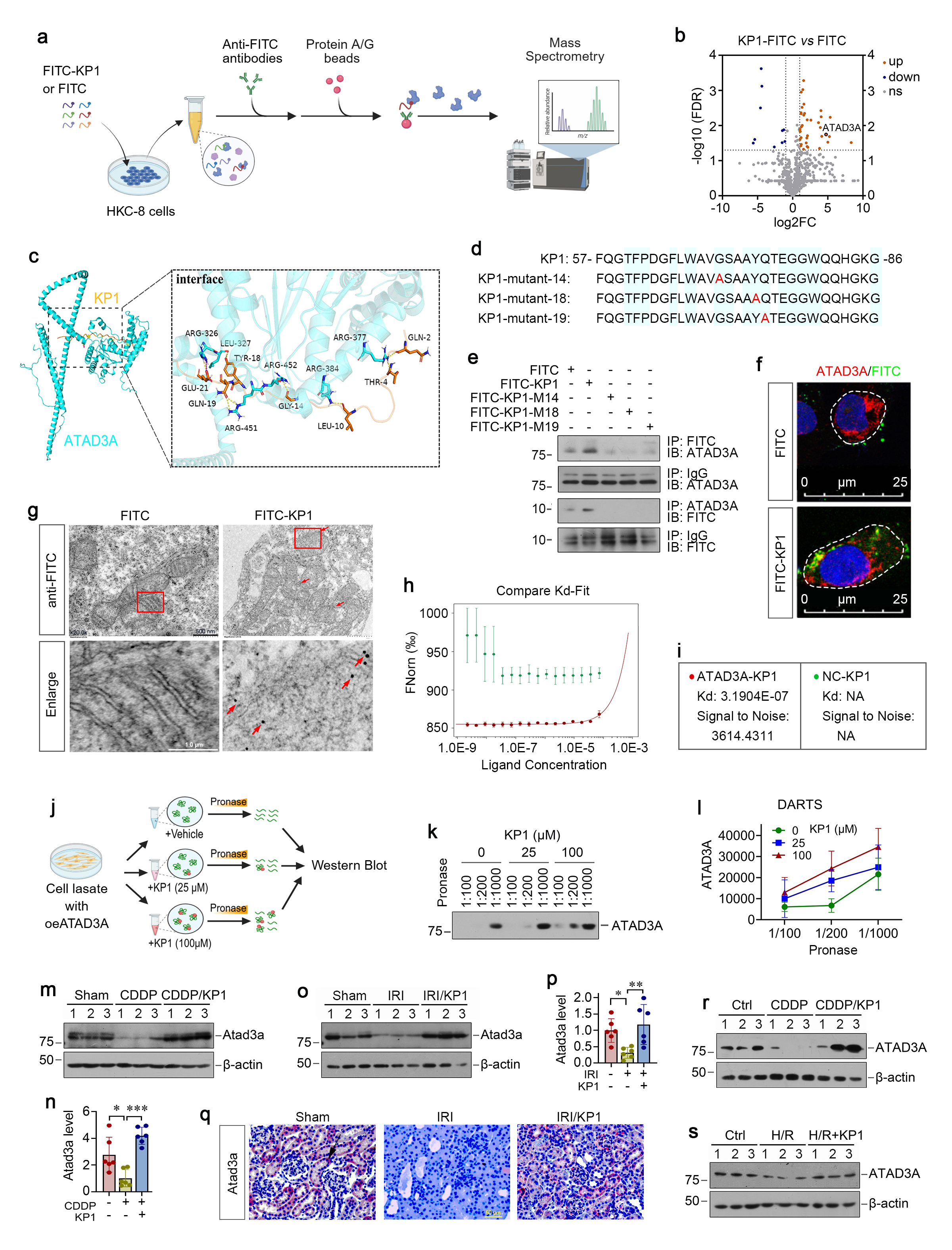
To explore the role of ATAD3A in regulating AKI, we injected mice with expression plasmid encoding Flag-tagged ATAD3A (pFlag-ATAD3A) 2 days before injection with CDDP. As shown in Figure 5a and b, overexpression of exogenous ATAD3A reduced Scr and BUN levels and mitigated kidney dysfunction. Notably, the expression of exogenous ATAD3A was confirmed by immunoblotting for Flag and ATAD3A in the kidneys (Figure 5c and e). Immunohistochemical staining also demonstrated that ATAD3A expression was completely restored in renal tubular epithelium in mice injected with pFlag-ATAD3A plasmid (Figure 5d). Overexpression of exogenous ATAD3A also suppressed the expression of tubular injury markers Kim-1, NGAL (Fig. 5c, e) and apoptosis markers PARP-1, cleaved caspase-3/9 (Fig. 5f and g). TUNEL staining and histopathological assessment revealed a reduced TUNEL⁺ cells, accompanied by mitigated tubular dilation and cast deposition after ATAD3A overexpression (Fig. 5d). ATAD3A also preserved mitochondrial proteins OPA1, Sam50 and mitofilin in the kidney after CDDP treatment (Fig. 5g-h).
We also investigated the effect of exogenous ADAT3A on IRI. As shown in Figure 5j-p, overexpression of ATAD3A decreased Scr and BUN levels, inhibited the expression of Kim-1, NGAL, PARP-1, p53 and cleaved caspase-3, and reduced TUNEL+ apoptotic cells in the kidneys of mice subjected to IRI. ATAD3A overexpression also largely preserved mitochondrial proteins OPA1, Sam50 and mitofilin, underscoring its role in protecting mitochondrial integrity (Figure 5 q and r).
ATAD3A alleviates tubular cell mitochondrial dysfunction after injury in vitro
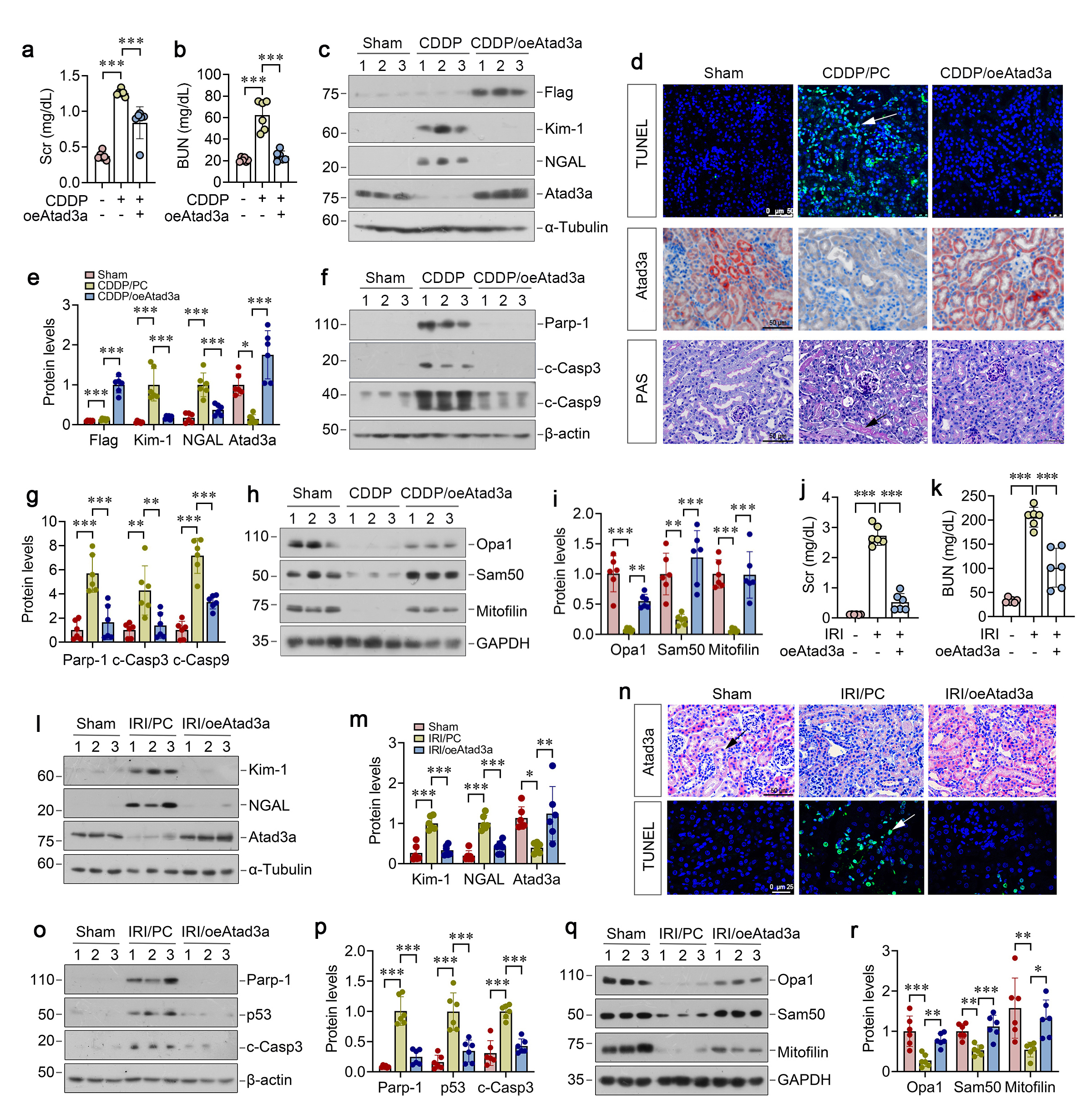
We further investigated the role of ATAD3A in protecting against mitochondrial dysfunction after injury by using cultured HKC-8 cells. As shown in Figure 6a and b, overexpressing ATAD3A restored its protein levels and suppressed Kim-1 and NGAL after CDDP treatment. Overexpression of ATAD3A also prevented CDDP-induced PARP-1, p53 and cleaved caspase-3 expression (Figure 6c and e). Live-time imaging confirmed that ATAD3A overexpression inhibited CDDP-triggered activation of caspase-3 and -7 (Figure 6e and f). The protective effect of ATAD3A was associated with the restoration of mitochondrial proteins OPA1, Sam50 and mitofilin in HKC-8 cells (Figure 6g and h), suggesting that ATAD3A alone sufficiently protects against CDDP-induced mitochondrial dysfunction and apoptosis.
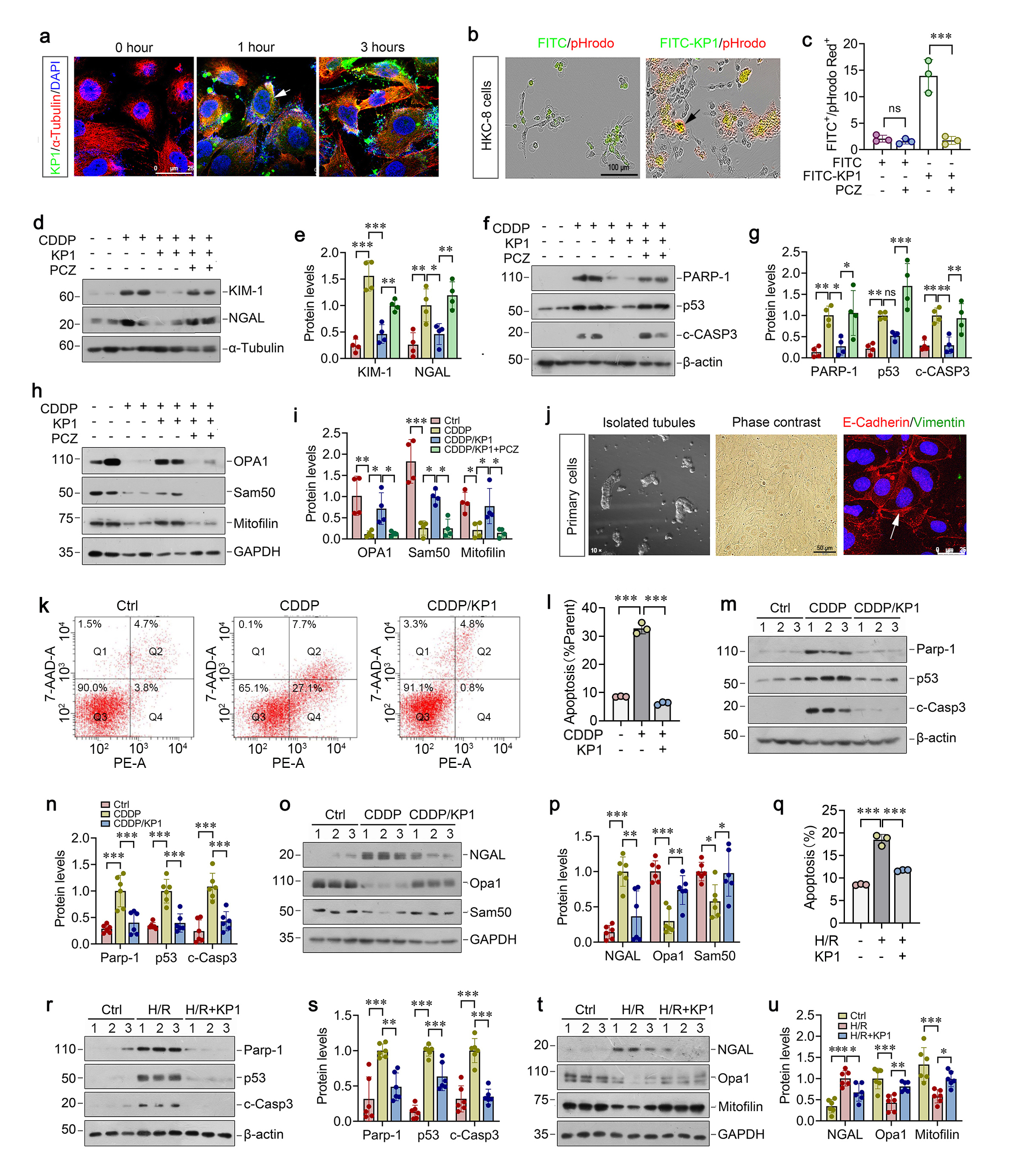
We then confirmed the role of ATAD3A in preserving mitochondrial integrity by knocking down its expression using small interfering RNA (siRNA) approach. As shown in Figure 6i-m, knockdown of ATAD3A by siATAD3A in HKC-8 cells slightly induced Kim-1, NGAL, PARP-1, p53, and cleaved caspase-3 expression, suggesting that ATAD3A depletion spontaneously induced tubular injury and apoptosis. Moreover, knockdown of ATAD3A further exacerbated CDDP-triggered induction of these proteins (Fig. 6i-m). ATAD3A loss also aggravated mitochondrial dysfunction, as evidenced by decreased OPA1, Sam50, and mitofilin proteins (Figure 6n and o).
ATAD3A preserves mitochondrial integrity through direct interaction with HIGD2A
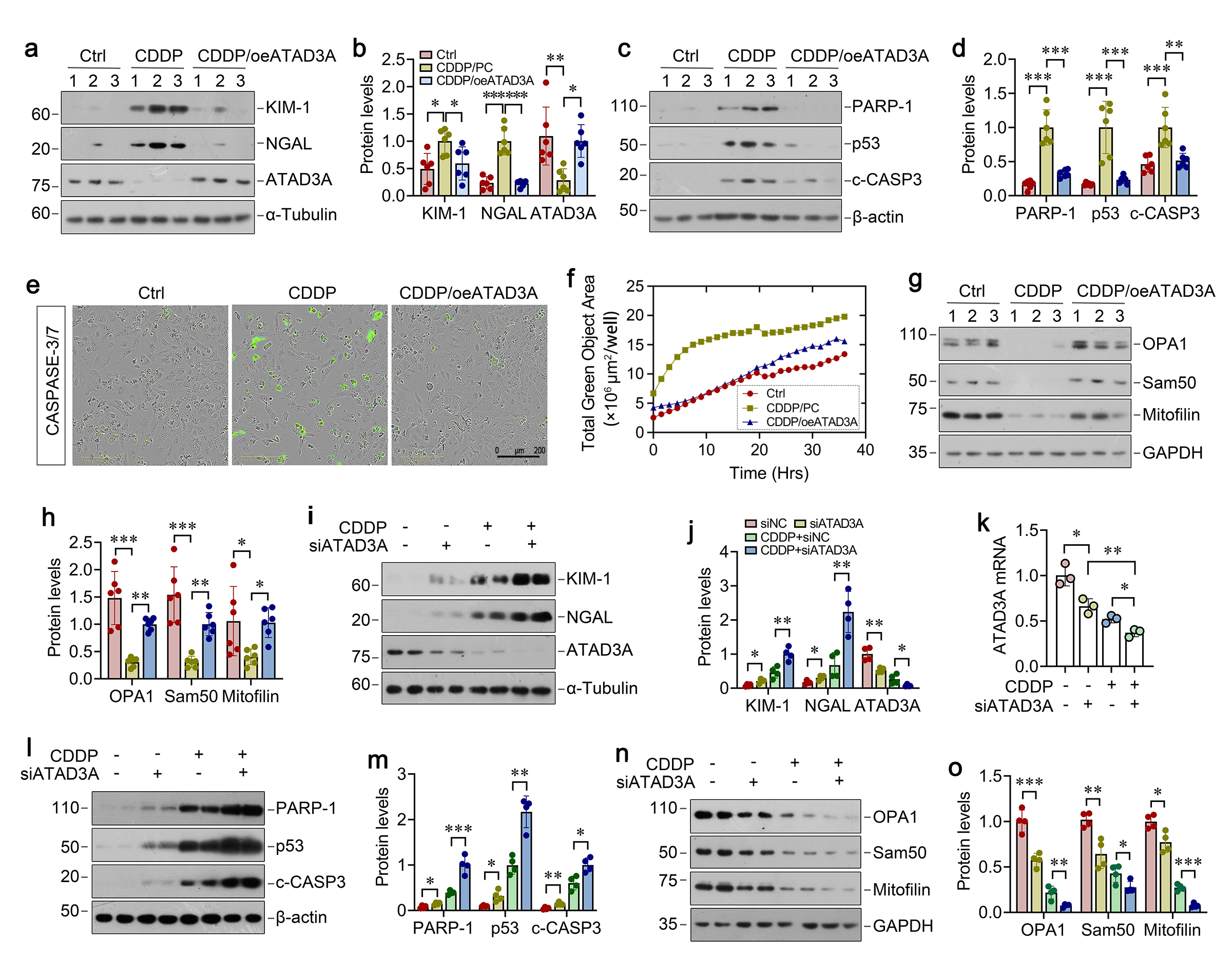
To elucidate the mechanism underlying ATAD3A protection against mitochondrial dysfunction, we sought to identify the direct targets of ATAD3A in mitochondria by using unbiased proteomic approach. To this end, mitochondria were isolated from kidneys and subjected to mass spectrometry (MS). We identified 58 upregulated and 17 downregulated proteins in IRI group versus sham controls (Figure 7a). Comparing to IRI, KP1 treatment resulted in 8 upregulated and 16 downregulated proteins (Figure 7a). Among them, KP1 restored the expression of 6 mitochondrial proteins that were downregulated in IRI kidneys. On the top of the list was the hypoxia inducible gene 1 (HIG1) domain family member 2A (HIGD2A), a subunit of the cytochrome c oxidase complex (complex IV) (Figure 7b). Molecular docking simulation predicted a strong ATAD3A-HIGD2A interaction, with a binding free energy of -115.99 kcal/mol (Figure 7c). To validate the interaction between ATAD3A and HIGD2A, we carried out co-immunoprecipitation assay. After incubation over-expressing Flag-tagged ATAD3A lysates with anti-HIGD2A antibody, ATAD3A was detected in the immunocomplexes (Figure 7d). In the reciprocal experiments, HIGD2A was also readily detected in the complexes precipitated by anti-Flag antibody (Figure 7d). Confocal microscopy confirmed the co-localization of FITC-KP1, ATAD3A, and HIGD2A in HKC-8 cells (Figure 7e).
We found that AKI caused by either CDDP or IRI resulted in marked downregulation of HIGD2A proteins, which was restored by KP1 in both models (Figure 7f-i). IHC also revealed that KP1 could restore HIGD2A expression in mice renal tubular with AKI induced by CDDP or IRI (Figure 7j and k). Similar results were obtained when HKC-8 cells were treated with CDDP or H/R (Figure 7l-o). We found that loss of HIGD2A after treatment with CDDP or H/R was associated with the release of cytochrome c from mitochondria to cytosol and KP1 reversed such cytochrome c translocation (Figure 7p and q), which could trigger tubular cell apoptosis.
HIGD2A protects against renal tubular cell apoptosis
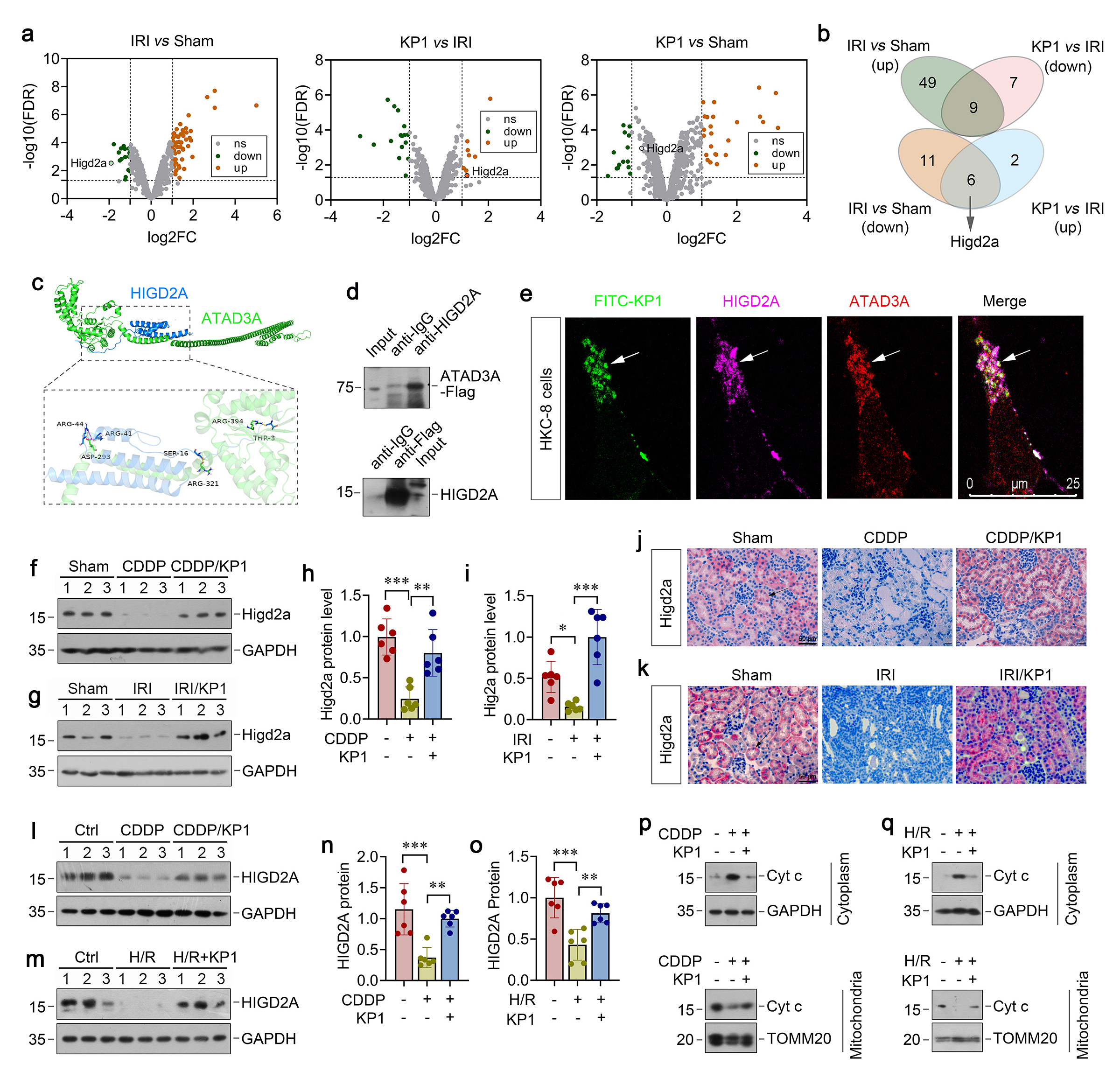
To confirm the role of HIGD2A in protecting against tubular cell apoptosis, we manipulated the expression of HIGD2A by knocking down or overexpression in HKC-8 cells in vitro. As shown in Figure 8a and b, knockdown of HIGD2A by siRNA aggravated CDDP-induced Kim-1 and NGAL expression in HKC-8 cells. HIGD2A depletion also worsened the loss of mitochondrial proteins OPA1, Sam50 and mitofilin induced by CDDP (Figure 7c and d). As a result, HIGD2A knockdown also aggravated PARP-1, p53, and cleaved caspase-3 expression induced by CDDP (Figure 8e-f), suggesting that HIGD2A deficiency further exacerbated tubular cell injury and apoptosis. Mitochondrial/cytosolic fractionation assays revealed that HIGD2A depletion promoted mitochondrial cytochrome c release into cytosol (Figure 8g).
We further investigated the role of HIGD2A in protecting against tubular cell apoptosis by overexpression. As shown in Figure 8h-m, overexpression of HIGD2A abolished the induction of Kim-1, NGAL PARP-1 p53 and cleaved caspase-9 and restored mitochondrial proteins OPA1, Sam50 and mitofilin in CDDP-treated HKC-8 cells, suggesting that HIGD2A protects against tubular injury, apoptosis, and mitochondrial dysfunction. Moreover, HIGD2A blocked CDDP-triggered cytochrome c release from mitochondria to cytosol (Fig. 8n), thereby leading to inhibition of downstream caspase-3 and -7 activation (Fig. 8o). Taken together, these results suggest that KP1 binds to mitochondrial protein ATAD3A, stabilizes ATAD3A/HIGD2A protein complex and prevents cytochrome c release from mitochondria to cytosol, thereby protecting tubular cells against apoptosis after injury (Figure 8p).